vcf_filter.py: VCF Filter Function¶
Depending on the analysis being conducted, a number of variant sites and/or samples may be unsuitable and must be removed. Given an unfiltered VCF and the desired filters, vcf_filter will apply the filters and produce a filtered VCF. Filters may be used independently or combined as needed. In addition, a number of the filters are seperated into two types: include (include/keep all relevant variant sites or samples) and exclude (exclude/remove all relevant variant sites or samples).
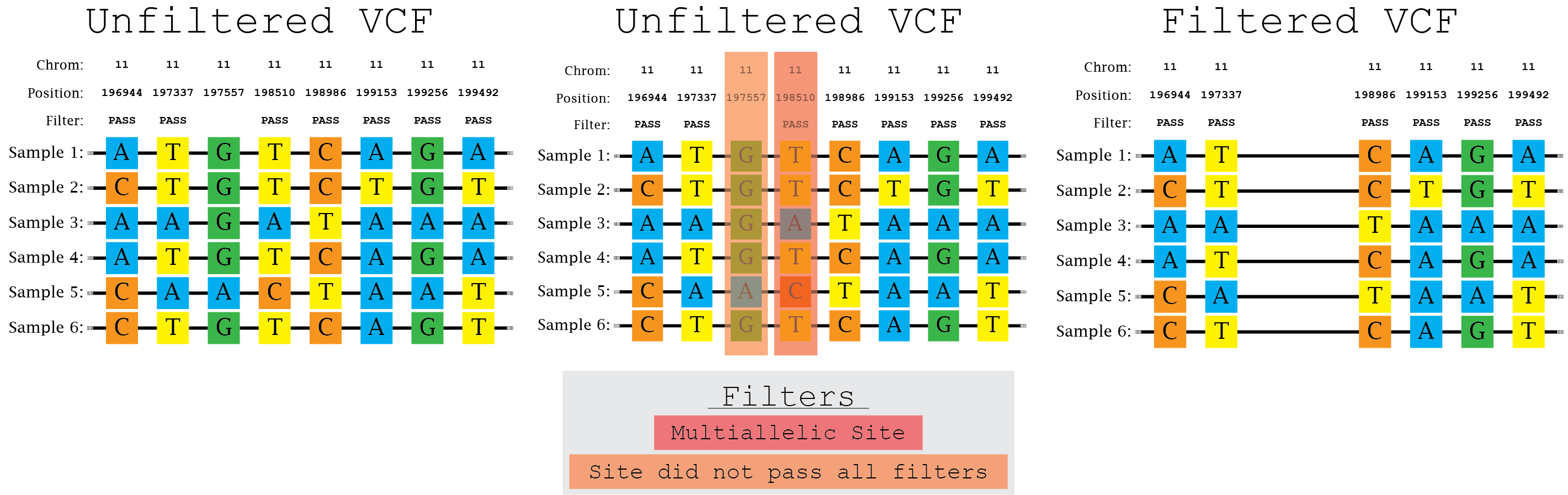
In this illustration of the filtering process (within a locus of interest), variant sites were kept only if they: i) were biallelic and ii) passed all filters. These requirements resulted in the removal of two variant sites (i.e. 197557 and 198510) within the given locus.
Command-line Usage¶
The VCF file filter may be called using the following command:
vcf_filter.py
Example usage¶
Command-line to create a BCF with only biallelic sites:
vcf_filter.py --vcf examples/files/merged_chr1_10000.vcf.gz --filter-only-biallelic --out-format bcf
Command-line to only include variants on chr1 from 1 to 1509546:
vcf_filter.py --vcf examples/files/merged_chr1_10000.bcf --filter-include-pos chr1:1-1509546
Command-line to remove indels and ouput a BCF file:
vcf_filter.py --vcf examples/files/merged_chr1_10000.indels.vcf.gz --filter-exclude-indels --out-format bcf
Input Command-line Arguments¶
- --vcf <input_filename>
- Argument used to define the filename of the VCF file to be filtered.
- --model-file <model_filename>
- Argument used to define the model file. Please note that this argument cannot be used with the individual-based filters.
- --model <model_str>
- Argument used to define the model (i.e. the individual(s) to include). Please note that this argument cannot be used with the individual-based filters.
Output Command-line Arguments¶
- --out <output_filename>
- Argument used to define the complete output filename, overrides --out-prefix
- --out-prefix <output_prefix>
- Argument used to define the output prefix (i.e. filename without file extension)
- --out-format <vcf, vcf.gz, bcf, bed, sites>
- Argument used to define the desired output format. Formats include: uncompressed VCF (vcf); compressed VCF (vcf.gz) [default]; BCF (bcf); variants in bed format; or variants in sites format.
- --overwrite
- Argument used to define if previous output should be overwritten.
Filter Command-line Arguments¶
The filtering arguments below are roughly seperated into catagoires. Please not that mulitple filters are seperated into two opposing function types include and exclude.
Individual-Based Arguments¶
Please note that all individual-based arguments are not compatible with either the --model or --model-file command-line arguments.
- --filter-include-indv <indv_str> <indv1_str, indv2_str, etc.>
- Argument used to define the individual(s) to include. This argument may be used multiple times if desired.
- --filter-exclude-indv <indv_str> <indv1_str, indv2_str, etc.>
- Argument used to define the individual(s) to exclude. This argument may be used multiple times if desired.
- --filter-include-indv-file <indv_filename>
- Argument used to define a file of individuals to include.
- --filter-exclude-indv-file <indv_filename>
- Argument used to define a file of individuals to exclude.
Allele/Genotype-Based Arguments¶
- --filter-only-biallelic
- Argument used to only include variants that are biallelic.
- --filter-min-alleles min_int
- Argument used to include variants with a number of allele >= to the given number.
- --filter-max-alleles max_int
- Argument used to include variants with a number of allele <= to the given number.
- --filter-maf-min maf_proportion
- Argument used to include variants with equal or greater MAF values.
- --filter-maf-max maf_proportion
- Argument used to include variants with equal or lesser MAF values.
- --filter-mac-min mac_int
- Argument used to include variants with equal or greater MAC values.
- --filter-mac-max mac_int
- Argument used to include variants with equal or lesser MAC values.
- --filter-include-indels
- Argument used to include variants if they contain an insertion or a deletion.
- --filter-exclude-indels
- Argument used to exclude variants if they contain an insertion or a deletion.
- --filter-include-snps
- Argument used to include variants if they contain a SNP.
- --filter-exclude-snps
- Argument used to exclude variants if they contain a SNP.
- --filter-include-snp <rs#> <rs#1, rs#2, etc.>
- Argument used to include SNP(s) with the matching ID. This argument may be used multiple times if desired.
- --filter-exclude-snp <rs#> <rs#1, rs#2, etc.>
- Argument used to exclude SNP(s) with the matching ID. This argument may be used multiple times if desired.
- --filter-include-snp-file <snp_filename>
- Argument used to define a file of SNP IDs to include.
- --filter-exclude-snp-file <snp_filename>
- Argument used to define a file of SNP IDs to exclude.
- --filter-max-missing proportion_float
- Argument used to filter positions by their proportion of missing data, a value of 0.0 allows for no missing whereas a value of 1.0 ignores missing data.
- --filter-max-missing-count count_int
- Argument used to filter positions by the number of samples with missing data, a value of 0 allows for no samples to have missing data.
Position-Based Arguments¶
- --filter-include-pos <chr, chr:pos, chr:start-end, chr:start->
- Argument used to include matching positions. May be used to include: an entire chromosome (i.e. chr); a single position (i.e. chr:pos); a chromosomal locus (i.e. chr:start-end); or a chromosomal span (i.e. chr:start-/chr:0-end). This argument may be used multiple times if desired.
- --filter-exclude-pos <chr, chr:pos, chr:start-end, chr:start->
- Argument used to exclude matching positions. May be used to exclude: an entire chromosome (i.e. chr); a single position (i.e. chr:pos); a chromosomal locus (i.e. chr:start-end); or a chromosomal span (i.e. chr:start-/chr:0-end). This argument may be used multiple times if desired.
- --filter-include-pos-file <position_filename>
- Argument used to define a file of positions to include within a tsv file (chromosome and position).
- --filter-exclude-pos-file <position_filename>
- Argument used to define a file of positions to exclude within a tsv file (chromosome and position).
- --filter-include-bed <position_bed_filename>
- Argument used to define a BED file of positions to include. Please note that filename must end in .bed.
- --filter-exclude-bed <position_bed_filename>
- Argument used to define a BED file of positions to exclude. Please note that filename must end in .bed.
Flag-Based Arguments¶
- --filter-include-passed
- Argument used to include positions with the 'PASS' filter flag.
- --filter-exclude-passed
- Argument used to exclude positions with the 'PASS' filter flag.
- --filter-include-filtered <filter_flag>
- Argument used to include positions with the specified filter flag.
- --filter-exclude-filtered <filter_flag>
- Argument used to exclude positions with the specified filter flag.
Other Command-line Arguments¶
- --force-samples
- Argument used to ignore the error rasied when a sample that does not exist within the input VCF.