informative_loci_filter.py: Informative Loci Filter¶
This function checks to make sure that a locus has enough sites to be considered informative in either the four-gamete test or an IM run. Given a BED file and a VCF file, informative_loci_filter will find regions in the VCF that have a specified number of variant sites.
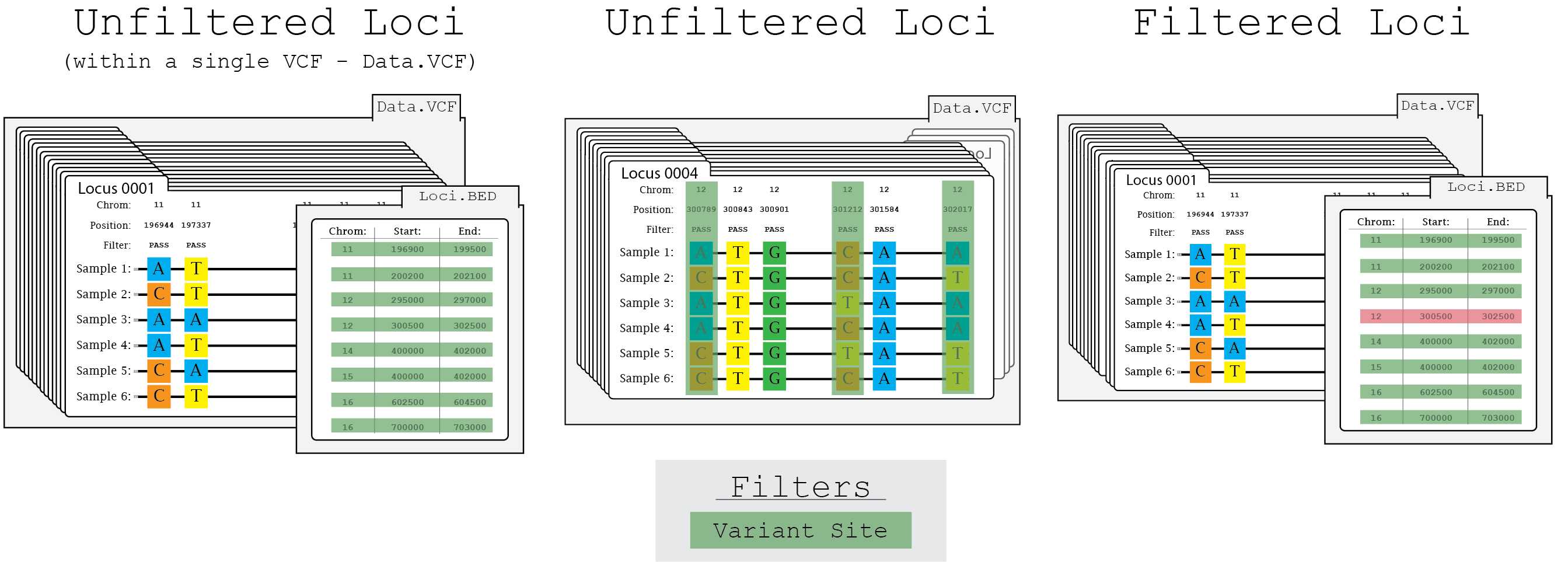
In this illustration of the locus filtering process, locus_0004 is removed due to only having three variant sites (highlighted in green) when the threshold is set to four.
Because many variants are not considered useful in these situations, filters are provided for removing sites with missing data, non-biallelic sites, indels, CpGs, and singletons from determining if there are a sufficient number of sites in the region. Output is either a BED file of a set number of random regions that pass the criteria, or a file with all regions that pass. It can also remove regions that are below a minimum specified length. If a model file is specified, only the individuals in the selected population will be considered for singleton and missing data filters.
Input Arguments¶
- --vcf <vcf_name>
- Name of input VCF file
- --bed <bed_file>
- Name of input BED file
Output Arguments¶
- --out <out_name>
- Name of output BED file
- --randcount <number_of_regions>
- If set, will output set number of regions randomly selected from those that pass the criteria. Default behavior is to output all regions that match criteria.
Filtering Arguments¶
- --remove-multi
- Do not count tri-allelic+ sites toward number of valid variants in a region
- --remove-missing <max_misscount>
- Do not count sites with more than max_misscount missing individuals. Default of -1 indicates all sites are included, 0 indicates sites with any missing data are not counted..
- --remove-indels
- Do not count indels toward number of valid variants in a region
- --parsecpg <fasta_reference_filename>
- Optional argument that if set, will detect whether or not variants are CpGs. A check is made to make sure the positions in the FASTA line up with the correct variant reference allele.
- --informative-count <minimum_allele_count>
- Minimum number of haplotypes with both alleles at a site. Default is 2, meaning there must be two of each of reference and alternate allele in the target individuals. Can be set to 1 to filter out invariant sites.
- --minsites <min_sites>
- Minimum number of variants required for a region to pass the filtering criteria. Variants that match specified arguments will not be counted towards this total.
- --min-length <min_length>
- Minimum base length of region for region to be considered.
Region Arguments¶
- --bed-column-index <start_idx>,*<end_idx>*,*<chrom_idx>*
- Comma-separated string of the zero-based indices of the start, end, and chromosome columns in the input file, so the file doesn't need to be reformatted. Default for a regular BED file is 1,2,0.
- --oneidx-start
If set, indicates input BED regions are formatted as one-indexed, closed intervals, as opposed to the BED default of zero-based, half-open intervals. For example, the first million bases on a chromosome would be:
Zero-based, half-open: 0,1000000 One-based, closed: 1,1000000- --pad <pad_length>
- If set, regions in input file will be extended by pad_length bases on both sides.
- --keep-full-line
- If set, regions output will be the same line as was present in the input file. Default behavior is to output start/end/chrom columns in that order, without any other data.
Model Arguments¶
- --model-file <model_filename>
- Name of model file that contains individuals to be considered for filtering.
- --model <model_name>
- If model file contains more than one model, name of model to be used.
Other Arguments¶
- --no-sorting
- Will output regions in order they were in input file. Default behavior is to sort regions before filtering.
- --tbi <tabix_index>
- If input VCF is compressed and tabix file is not default, provide the tabix filename here.
- --no-xy
- Removes regions on X/Y chromosomes from consideration.