bed_utilities.py: BED Utilites¶
Automates various utilites for BED-formatted files. This currently includes: i) sample a BED file; ii) subtract from a BED that overlap with a second BED file; iii) extend a BED upstream, downstream, or both upstream and downstream; iv) sort a single BED; v) merge features within one or more BED files; vi) create a BED of complementary features.
Command-line Usage¶
The BED utilites function may be called using the following command:
bed_utilities.py
Utilites¶
Windows Utility¶
Given a chromosome size file and a window size, the windows utility will generate a BED file of interval features.
Example usage¶
Return a BED with interval features that do not extend outside the chromosomes:
bed_utilities.py --utility windows --chrom-file hg18.chrom.sizes --window-size 1000 --out hg18_windows.bed
Sample Utility¶
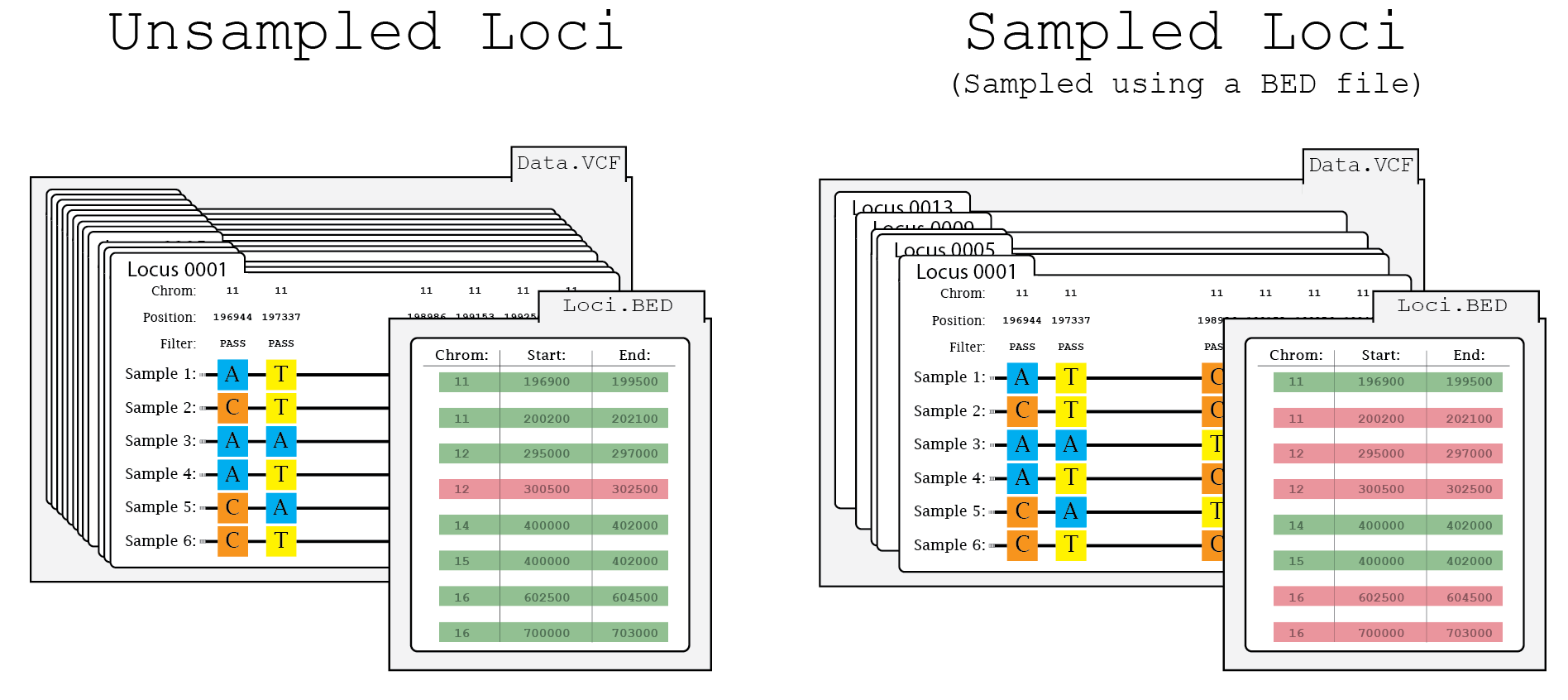
Given a BED file and a sample size, the sample utility will generate a pseudorandomly sampled BED. Please note that the random seed may be used to reproduced the sample.
Example usage¶
Sample 20 features from a BED file:
bed_utilities.py --utility sample --bed examples/files/chr1_sites.bed --sample-size 20
Sort Utility¶
Given an unsorted BED file, the sort utility will generate a sorted BED file.
Example usage¶
Sort an unsorted BED file:
bed_utilities.py --utility sort --bed examples/files/chr1_sites.unsorted.bed
Extend Utility¶
Given a BED file and an extend length, the extend utility will increase the length of each feature upstream, downstream, or both upstream and downstream.
Example usage¶
Extend upstream by 1kb:
bed_utilities.py --utility extend --bed examples/files/chr1_sites.bed --chrom-file examples/files/chr_sizes.txt --extend-upstream 1000
Extend downstream by 1kb:
bed_utilities.py --utility extend --bed examples/files/chr1_sites.bed --chrom-file examples/files/chr_sizes.txt --extend-downstream 1000
Extend flanks (i.e. both upstream and downstream) by 1kb:
bed_utilities.py --utility extend --bed examples/files/chr1_sites.bed --chrom-file examples/files/chr_sizes.txt--extend-flanks 1000
Subtract Utility¶
Given two BED files, the subtract utility will remove BED features from a BED file if they overlap with the features from a second BED file.
Example usage¶
Remove BED features if they overlap features within the subtract BED file:
bed_utilities.py --utility subtract --bed examples/files/chr1_sites.bed --subtract-bed examples/files/chr1_sites.1.bed --subtract-entire-feature
Complement Utility¶
Given a BED file, the complementary utility will generate a BED file of complementary features.
Example usage¶
Return a BED with features that do not overlap within the given file:
bed_utilities.py --utility complement --bed examples/files/chr1_sites.bed --chrom-file examples/files/chr_sizes.txt
Intersect Utility¶
Given a BED file and an intersect file, return only the interval features within the BED file that overlap with the intersect file.
Example usage¶
Return a BED with only intersecting interval features:
bed_utilities.py --utility intersect --bed hg18_windows.bed --intersect-file Intersect.vcf.gz --out hg18_intersects.bed
Merge Utility¶
Given one or more BED files, the merge utility will generate a single sorted BED file of merged BED features.
Example usage¶
Merge BED features from a single BED file:
bed_utilities.py --utility merge --bed examples/files/chr1_sites.bed
Merge BED features from multiple BED files:
bed_utilities.py --utility merge --beds examples/files/chr1_sites.1.bed examples/files/chr1_sites.2.bed examples/files/chr1_sites.3.bed examples/files/chr1_sites.4.bed
Input Command-line Arguments¶
- --bed <input_filename>
- Argument used to define the filename of the BED file.
- --beds <input_filename> <input1_filename, input2_filename, etc.>
- Argument used to define the filename of the BED file(s). May be used multiple times.
- --chrom-file <chrom_filename>
Argument used to define the filename of a file with the sizes of each chromosome. Chromosome size files must be tab-delimited as follows:
chr1 247249719 chr2 242951149 ... chrX 154913754 chrY 57772954
Appropriate files may be downloaded from the UCSC Genome Browser. The supported ASSEMBLY.chrom.sizes file for each assembly may be found by clicking Genome sequence files and select annotations (followed by Standard genome sequence files and select annotations on select assemblies).
Output Command-line Arguments¶
- --out <output_filename>
- Argument used to define the complete output filename.
- --overwrite
- Argument used to define if previous output should be overwritten.
Utility Command-line Specification¶
- --utility <sample, subtract, extend, sort, merge, complement>
- Argument used to define the desired utility. Current utilities include: sample features from a BED file (sample); subtract features from a BED file that overlap with features within a second BED file (subtract); extend the flanks of features upstream, downstream, or both within a single BED file (extend); sort the features within a single BED file (sort); merge features within one or more BED files (merge); create a BED file of complementary features - i.e. features that do not overlap - from a BED file (complement).
Window Utility Command-line Arguments¶
- --window-size <window_size_int>
- Argument used to define the window/interval size to return.
Sample Utility Command-line Arguments¶
- --sample-size <sample_size_int>
- Argument used to define the total sample size.
- --random-seed <seed_int>
- Argument used to define the seed value for the random number generator.
Subtract Utility Command-line Arguments¶
- --subtract-bed <subtract_file_filename>
- Argument used to define the BED file used for removing features/positions.
- --subtract-entire-feature
- Argument used to define if entire features within the input BED should be removed if they overlap with features in subtract-bed.
- --min-reciprocal-overlap <overlap_float>
- Argument used to define the minimum reciprocal overlap of features required for removal (e.g. 0.1 indicates 10% overlap).
- --min-input-overlap <overlap_float>
- Argument used to define the minimum overlap of input features required for removal.
- --min-subtract-overlap <overlap_float>
- Argument used to define the minimum overlap of subtract-bed features required for removal.
- --subtract-entire-feature
- Argument used to define that features should be removed from the input BED if the minimum overlap of --min-input-overlap or --min-subtract-overlap is reached.
Extend Utility Command-line Arguments¶
- --extend-flanks <bp_int>
- Argument used to define the length of base pairs (bp) to extend both upstream and downstream of features.
- --extend-upstream <bp_int>
- Argument used to define the length of base pairs (bp) to extend upstream of features.
- --extend-downstream <bp_int>
- Argument used to define the length of base pairs (bp) to extend downstream of features.
Intersect Utility Command-line Arguments¶
- --intersect-file <intersect_file_filename>
- Argument used to define the BED/VCF/VCF.gz file used to remove features that do not intersect with the given file's features/variants. removing features/positions.
Merge Utility Command-line Arguments¶
- --max-merge-distance <bp_int>
- Argument used to define the maximum distance allowed between features to be merged.